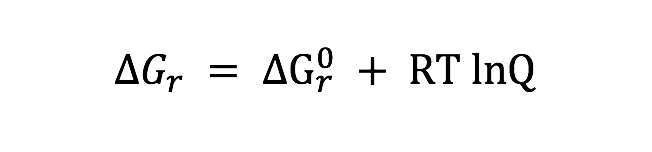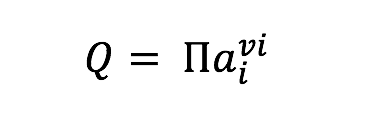Tables and Figures referenced in the acquisition description are found in the paper Frank et al., 2015
Once on board ship, tubeworms and other macrofauna were removed from the samples and the large pieces were broken into more manageable fragments (~10-20 cm3) with a flame-sterilized chisel and sledgehammer, with the user wearing sterile nitrile gloves. Samples were quickly transferred to 0.2 um-filtered anaerobic (nitrogen-sparged) seawater. Samples were further broken down into smaller sizes while in anaerobic water, and subsamples from the interior of the fragments were immediately transferred to gastight jars (Freund Container Inc.) filled with sterile anaerobic seawater containing 2 mM sodium sulfide at pH 6, and stored at 4 degrees celsius for incubations and analyses. The sterile sulfidic seawater in the gastight jars were refreshed periodically during storage at 4 degrees celsius. The majority of the rate experiments (80%) were set up immediately on the ship using freshly collected samples. In parallel, subsamples (~1 cm3) from each flange were preserved aboard ship in glutaraldehyde (2.5% in phosphate buffered saline, PBS, pH 7.0), then prepared for electron microscopy via ethanol dehydration and critical point drying before being sputtered with a thin layer of gold-palladium to improve image resolution. Samples were imaged with a Zeiss model EVO Scanning Electron Microscope (SEM).
Prior to incubation, each flange subsample was pulverized by hand for about one hour to minimize fine-scale geological and microbial heterogeneity and facilitate more accurate experimental replication (akin to slurry experiments in sediments; Fossing & Jørgensen 1989; Weber & Jørgensen 2002; Jørgensen et al. 1992). Specifically, each subsample was pulverized with a flame-sterilized sledgehammer in sterile seawater actively bubbled with nitrogen within an anaerobic chamber. For each independent treatment, aliquots of 7.5 mL flange slurry (approx. 29 g wet weight and 20 g dry weight) were transferred into Balch tubes in an anaerobic chamber, and supplemented with 15 mL of sterile artificial seawater media designed to mimic the geochemical conditions within a hydrothermal flange (400 mM NaCl, 25 mM KCl, 30 mM CaCl2, 2.3 mM NaHCO3, 14 mM NaSO42-, 1 mM H2S, and 50 uM dissolved organic carbon – consisting of equimolar proportions 10 uM of pyruvate, citrate, formate, acetate, lactate) under a pure nitrogen headspace.
Concentrations of sulfide, sulfate and dissolved organic carbon (DOC) were varied independently to investigate concentration dependent effects on the rates of SR. The range of experimental conditions tested was determined from previously published concentration profiles of aqueous species modeled as functions of temperature and position within the Grotto vent structure (Tivey, 2004). Concentrations were varied by orders of magnitude within the modeled ranges to simulate conditions representative of different mixing regimes between seawater and vent fluid (Table 1). The range of DOC (which we approximate as a mix of pyruvate, citrate, formate, acetate, lactate – most of which have been identified to varying degrees within vent fluid and are known carbon sources for heterotrophic SR in culture) concentrations tested were based on the average DOC concentrations measured within diffuse fluids at the Main Endeavor Field (Lang et al., 2006; Lang et al., 2010). Hydrogen sulfide was present as H2S (pKa in seawater of 6.60) across all the conditions tested (Amend & Shock, 2001). Incubations were carried out at pH 4 (to simulate the pH of end-member Grotto vent fluid and the average calculated pH of mixed fluids in highly reduced zones within the flange; Tivey 2004) as well as pH 6 (representative of the calculated pH in fluid mixing zones; Tivey 2004). All the results are presented and discussed in the context of the initial measured media conditions.
Sufficient 35SO42- was added to achieve 15 uCi of activity. Samples were incubated anaerobically for 1, 3 or 7 days at ambient seawater (4 degrees celsius), thermophilic (50 degrees celsius) and hyperthermophilic (90 degrees celsius) temperatures. The range of temperatures considered was representative of different thermal regimes associated with the surface, outer layer and middle regions of hydrothermal chimneys (Tivey 2004; Kormas et al. 2006; Schrenk et al. 2003). Negative controls consisted of samples amended with 28 mM molybdate to inhibit SR (Newport & Nedwell, 1988; Saleh et al., 1964). Three biological replicates were run for each treatment, and two biological replicates for each control.
Upon completion, reactions were quenched with the injection of 5 mL 25% zinc acetate, at pH 8 (i.e. 20-fold excess Zn), and all samples were frozen at -20 degrees celsius for further analysis. 80% of incubations were performed shipboard with freshly collected samples and the remaining 20% of incubations were completed within one year of collection.
To determine SR rates, samples were thawed and the supernatant was removed and filtered through a 0.2 um syringe filter. The homogenized flange that remained in the tube was washed three times with deionized water to remove any remaining sulfate. One gram (wet weight) of flange material was added to 10 mL of a 1:1 ethanol to water solution in the chromium distillation apparatus, and then degassed with nitrogen for 15 minutes to drive the environment anoxic. Hydrogen sulfide gas was evolved after the anaerobic addition of 8 mL of 12 N HCl and 10 mL of 1 M reduced chromium chloride, followed by 3 hours of heating. The resulting hydrogen sulfide gas was carried via nitrogen gas through a condenser to remove HCl, and was then trapped as zinc sulfide in a 25% zinc acetate solution. To moderate potential artifacts of hot distillation methods including elevated rates in control samples, experiments were analyzed in triplicate, on different days and with different glassware to minimize cross-contamination, and any activity observed in “control” samples was deleted from the treatments. The radioactivity of the resulting sulfide (Zn35S) and the remaining sulfate from the supernatant (35SO42-) were measured via liquid scintillation counter in Ultima Gold scintillation cocktail (ThermoFisher Inc., Waltham, MA).
Once on board ship, tubeworms and other macrofauna were removed from the samples and the large pieces were broken into more manageable fragments (~10-20 cm3) with a flame-sterilized chisel and sledgehammer, with the user wearing sterile nitrile gloves. Samples were quickly transferred to 0.2 um-filtered anaerobic (nitrogen-sparged) seawater. Samples were further broken down into smaller sizes while in anaerobic water, and subsamples from the interior of the fragments were immediately transferred to gastight jars (Freund Container Inc.) filled with sterile anaerobic seawater containing 2 mM sodium sulfide at pH 6, and stored at 4 degrees celsius for incubations and analyses. The sterile sulfidic seawater in the gastight jars were refreshed periodically during storage at 4 degrees celsius. The majority of the rate experiments (80%) were set up immediately on the ship using freshly collected samples. In parallel, subsamples (~1 cm3) from each flange were preserved aboard ship in glutaraldehyde (2.5% in phosphate buffered saline, PBS, pH 7.0), then prepared for electron microscopy via ethanol dehydration and critical point drying before being sputtered with a thin layer of gold-palladium to improve image resolution. Samples were imaged with a Zeiss model EVO Scanning Electron Microscope (SEM).
Prior to incubation, each flange subsample was pulverized by hand for about one hour to minimize fine-scale geological and microbial heterogeneity and facilitate more accurate experimental replication (akin to slurry experiments in sediments; Fossing & Jørgensen 1989; Weber & Jørgensen 2002; Jørgensen et al. 1992). Specifically, each subsample was pulverized with a flame-sterilized sledgehammer in sterile seawater actively bubbled with nitrogen within an anaerobic chamber. For each independent treatment, aliquots of 7.5 mL flange slurry (approx. 29 g wet weight and 20 g dry weight) were transferred into Balch tubes in an anaerobic chamber, and supplemented with 15 mL of sterile artificial seawater media designed to mimic the geochemical conditions within a hydrothermal flange (400 mM NaCl, 25 mM KCl, 30 mM CaCl2, 2.3 mM NaHCO3, 14 mM NaSO42-, 1 mM H2S, and 50 uM dissolved organic carbon – consisting of equimolar proportions 10 uM of pyruvate, citrate, formate, acetate, lactate) under a pure nitrogen headspace.
Concentrations of sulfide, sulfate and dissolved organic carbon (DOC) were varied independently to investigate concentration dependent effects on the rates of SR. The range of experimental conditions tested was determined from previously published concentration profiles of aqueous species modeled as functions of temperature and position within the Grotto vent structure (Tivey, 2004). Concentrations were varied by orders of magnitude within the modeled ranges to simulate conditions representative of different mixing regimes between seawater and vent fluid (Table 1). The range of DOC (which we approximate as a mix of pyruvate, citrate, formate, acetate, lactate – most of which have been identified to varying degrees within vent fluid and are known carbon sources for heterotrophic SR in culture) concentrations tested were based on the average DOC concentrations measured within diffuse fluids at the Main Endeavor Field (Lang et al., 2006; Lang et al., 2010). Hydrogen sulfide was present as H2S (pKa in seawater of 6.60) across all the conditions tested (Amend & Shock, 2001). Incubations were carried out at pH 4 (to simulate the pH of end-member Grotto vent fluid and the average calculated pH of mixed fluids in highly reduced zones within the flange; Tivey 2004) as well as pH 6 (representative of the calculated pH in fluid mixing zones; Tivey 2004). All the results are presented and discussed in the context of the initial measured media conditions.
Sufficient 35SO42- was added to achieve 15 uCi of activity. Samples were incubated anaerobically for 1, 3 or 7 days at ambient seawater (4 degrees celsius), thermophilic (50 degrees celsius) and hyperthermophilic (90 degrees celsius) temperatures. The range of temperatures considered was representative of different thermal regimes associated with the surface, outer layer and middle regions of hydrothermal chimneys (Tivey 2004; Kormas et al. 2006; Schrenk et al. 2003). Negative controls consisted of samples amended with 28 mM molybdate to inhibit SR (Newport & Nedwell, 1988; Saleh et al., 1964). Three biological replicates were run for each treatment, and two biological replicates for each control.
Upon completion, reactions were quenched with the injection of 5 mL 25% zinc acetate, at pH 8 (i.e. 20-fold excess Zn), and all samples were frozen at -20 degrees celsius for further analysis. 80% of incubations were performed shipboard with freshly collected samples and the remaining 20% of incubations were completed within one year of collection.
To determine SR rates, samples were thawed and the supernatant was removed and filtered through a 0.2 um syringe filter. The homogenized flange that remained in the tube was washed three times with deionized water to remove any remaining sulfate. One gram (wet weight) of flange material was added to 10 mL of a 1:1 ethanol to water solution in the chromium distillation apparatus, and then degassed with nitrogen for 15 minutes to drive the environment anoxic. Hydrogen sulfide gas was evolved after the anaerobic addition of 8 mL of 12 N HCl and 10 mL of 1 M reduced chromium chloride, followed by 3 hours of heating. The resulting hydrogen sulfide gas was carried via nitrogen gas through a condenser to remove HCl, and was then trapped as zinc sulfide in a 25% zinc acetate solution. To moderate potential artifacts of hot distillation methods including elevated rates in control samples, experiments were analyzed in triplicate, on different days and with different glassware to minimize cross-contamination, and any activity observed in “control” samples was deleted from the treatments. The radioactivity of the resulting sulfide (Zn35S) and the remaining sulfate from the supernatant (35SO42-) were measured via liquid scintillation counter in Ultima Gold scintillation cocktail (ThermoFisher Inc., Waltham, MA).
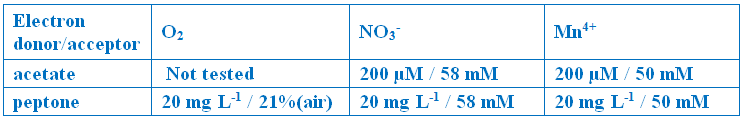 Saline basal medium was composed of (g/L): 0.2 NH4Cl, 30 NaCl, 2.8 MgCl2, 0.33 KCl, 0.3 CaCl2, 0.3 KH2PO4, 0.01 NaBr, 0.015 H3BO3, 0.02 SrCl2, 0.02 KI, 0.02 FeCl3, 0.0075 MnSO4, 0.0045 Na2WO4.2H2O, 0.003 NiCl2, 0.02 CoSO4, 0.0015 ZnSO4, 0.002 CuSO4, and 0.0015 Na2MoO4. pH was adjusted to 6.2.
Saline basal medium was composed of (g/L): 0.2 NH4Cl, 30 NaCl, 2.8 MgCl2, 0.33 KCl, 0.3 CaCl2, 0.3 KH2PO4, 0.01 NaBr, 0.015 H3BO3, 0.02 SrCl2, 0.02 KI, 0.02 FeCl3, 0.0075 MnSO4, 0.0045 Na2WO4.2H2O, 0.003 NiCl2, 0.02 CoSO4, 0.0015 ZnSO4, 0.002 CuSO4, and 0.0015 Na2MoO4. pH was adjusted to 6.2.